*The picture in the header was taken from here.
This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison.
______________________________________________________________________________________________________
Conclusion
The purpose of this project is to explore how UBE3A gene affects Angelman Syndrome (AS) patients via various online bioinformatics tool. I found that UBE3A is conserved in organisms such as chimpanzee, gray wolf, cattle, mouse, rat, chicken, zebrafish, fruit fly, and mosquito. Organisms that possess UBE3A gene homologs can be useful model organisms for research purposes, especially mouse since they are easy to manage and have a moderate life cycle.
I began to search for UBE3A gene ontology and most of them are related to protein modification via ubiquitination. I look up several protein domain databases and all of them showed that E6AP (the protein encoded by UBE3A) only has a domain: the HECT domain, which plays a role in ubiquitination. By looking at the protein interaction network generated by STRING, I found that before E6AP transfers the ubiquitin molecule to targeted protein molecules, it receives ubiquitin in the form of a thioester from E2 ubiquitin-conjugating enzyme.
There are four main causes of AS but they are all due to the fact that the paternal UBE3A allele is silenced is neurons. Although UBE3A is expressed in many tissues, it is only in neurons that the paternal allele is not active. The four main causes are deletion of the maternal UBE3A gene, uniparental disomy (UPD) whereby the patient inherited two chromosomes 15 from the father and both UBE3A alleles are silenced, defect in the imprinting center which silences the maternal UBE3A and lastly, mutation in the UBE3A gene itself.
I was wondering why the paternal allele is silenced only in neurons and further investigation showed that the genomic imprinting is regulated by Prader-Willi Syndrome Imprinting Center (PWS-IC) and Angelman Syndrome Imprinting Center (AS-IC). PWS-IC is a 300 bp segment at chromosome 15q11-q13. It is regarded as the master regulator of imprinting of the chromosome 15q11-q13 region and has a CG-rich cluster. The CG rich region of the paternal copy of PWS-IC is not methylated and therefore, it is active and allows the transcription of a noncoding antisense RNA (UBE3A-ATS). On the other hand, the maternal PWS-IC is methylated and thus there is no transcription of UBE3A-ATS. UBE3A-ATS is proposed to be responsible for the silencing of the paternal UBE3A allele but the mechanism is still unknown. Further upstream of the PWS-IC is the AS-IC, which is 880bp long. It was observed that if the AS-IC is activated, the PWS-IC is methylated and therefore, is silenced.
Then, I started to ask how AS-IC is imprinted that led to the eventual imprinting of PWS-IC and UBE3A. I looked at the protein interaction network again and found that SIN3A is a transcriptional repressor protein. This prompted me to ask the following questions:
1) Does SIN3A involve in the silencing of the paternal UBE3A?
2) Does it interact with the AS-IC?
3) Is SIN3A only expressed paternally in neurons?
I extended the protein network to see what other proteins SIN3A interacts with and found that it interacts with MECP2, HDAC1 and HDAC2. With further readings, I understand that MECP2 first binds to methylated DNA and later recruits the SIN3A/HDAC complex. The HDAC complex will then remove the acetyl groups from histones, causing the remodeling of the chromatin structure and silencing of the gene in that region. Therefore, I came up with the hypothesis that MECP2 recruits the SIN3A/HDAC complex and changes the structure of the chromatin. In this experiment, I proposed that mouse hippocampal neurons are used for several reasons:
1) SIN3A in mouse and in human are 97% identical, as shown by BLAST.
2) Neurons are chosen for the obvious reason that UBE3A is only silenced in neurons.
3) Hippocampal neurons are selected among other neurons because it was found that UBE3A is highly expressed in hippocampus.
I would then use Tandem Affinity Purification tag (TAP tag) and target the SIN3A protein. After that, I would perform a mass spectrometry to identify these molecules. Since SIN3A interacts with MECP2 and the HDAC complex, I would expect to identify these proteins.
Next, I would try to find which part of the chromosome this complex interacts with. I hypothesized that it would most likely be the AS-IC because it is known that the inactivation of the AS-IC activates the PWS-IC, which allows the expression of UBE3A-ATS. Therefore, this complex would bind to the AS-IC and inactivates it, which leads to the eventual silencing of UBE3A. To address this, I would perform a ChIP assay on mice hippocampal neurons, amplify the DNA fragment via PCR and finally sequence this region. Since we know that the AS-IC is 880bp long, we would expect a band around the 800-900bp region. To further verify the identity of this DNA fragment, I would perform a BLAST search by using the sequencing data.
Lastly, I hypothesized that SIN3A is only expressed paternally in neurons. The rationale behind this is that the expression of the paternal SIN3A causes the inactivation of the paternal AS-IC, which eventually silences the paternal UBE3A allele. To test this, I proposed to conduct an experiment using transgenic mice with YFP gene. In this experiment, there will be 3 strains of mice: Wildtype (+/+), SIN3AYFP paternal (+/YFP) and SIN3AYFP maternal (YFP/+). First, I would produce mice with a knock-in mutation to fuse YFP to Sin3a and later cross the WT mice with the SIN3AYFP mice to obtain offspring that inherit the SIN3AYFP allele either paternally or maternally. Then I would use western blotting to determine which Sin3a allele is of which parent of origin. SIN3A:YFP protein is bigger than Sin3a protein and with this information, if the SIN3AYFP allele is inherited paternally, we would expect an increase in the molecular weight of the Sin3a protein. However, if the SIN3AYFP allele is inherited maternally, this allele will not be expressed and we will see a band that is similar to that of the wild type control.
Besides that, we can also use confocal imaging to look at the localization of the protein in the brain. The brain sections would be immunostained with anti-YFP and we would expect SIN3A to be highly expressed in the hippocampus.
In conclusion, if this hypothesis stands true, then the paternal SIN3A allele is expressed, which causes the paternal AS-IC to be inactivated, the paternal PWS-IC to be activated, and the paternal UBE3A-ATS to be expressed, which leads to the silencing of the paternal UBE3A allele.
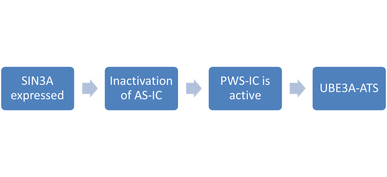
Figure 1: If my hypothesis stands true, the expression of paternal SIN3A eventually causes the silencing of the paternal UBE3A.
Future Directions
1. Analysis of SIN3A domains and functions
By looking at SIN3A domains, we would be able to tell what other functions SIN3A has and what other proteins that SIN3A interacts with have these domains.
2. Identify inhibitors of SIN3A
I would use PubChem to search for inhibitors of SIN3A and test to see if these inhibitors can activate the paternal AS-IC.
3. Identify the genes in AS-IC
I would first try to find the genes in this region via BLAST and later look at each gene’s function. Then, I would try to correlate how these genes could affect neurons/synapses formation and also AS patients.
References:
[1] AMIGO
[2] Pfam
[3] SMART
[4] STRING 9.0
[5] Angelman Syndrome Foundation
[6] Chamberlain, S. J., Lalande, M. (2010). Angelman Syndrome, a Genomic Imprinting Disorder of the Brain. The Journal of Neuroscience, 30(30):9958 –9963. doi:10.1523/jneurosci.1728-10.2010.
[7] Mabb, A. M., Judson, M. C., Zylka, M. J., Philpot, B. D. (2011). Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends in Neurosciences, 34(6): 293-303. doi: 10.1016/j.tins.2011.04.001.
[8] Dindot, S. V., Antalffy, B. A., Bhattacharjee, M. B., Beaudet, A. L. (2007). The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Human Molecular Genetics, 17(1): 111-118. doi:10.1093/hmg/ddm288
______________________________________________________________________________________________________
If you find my website helpful, please consider donating to the Foundation for Angelman Syndrome Therapeutics (FAST)

Created by Jonathan Mok
[email protected]
Last updated 05/06/2012
Genetics 677

Created by Jonathan Mok
[email protected]
Last updated 05/06/2012
Genetics 677
